Writing molecular dynamics data to binary LAMMPS format
In this post I will explain how to write to the binary LAMMPS file format from C++, using data stored in Armadillo vectors and matrices. After running the example in this post you should be able to open the resulting file in Ovito or any other program capable of reading binary LAMMPS files. The example should also be fairly easy to port to other data structure type, if needed.
For the impatient: You’ll find a working main.cpp file and a qmake project file on GitHub.
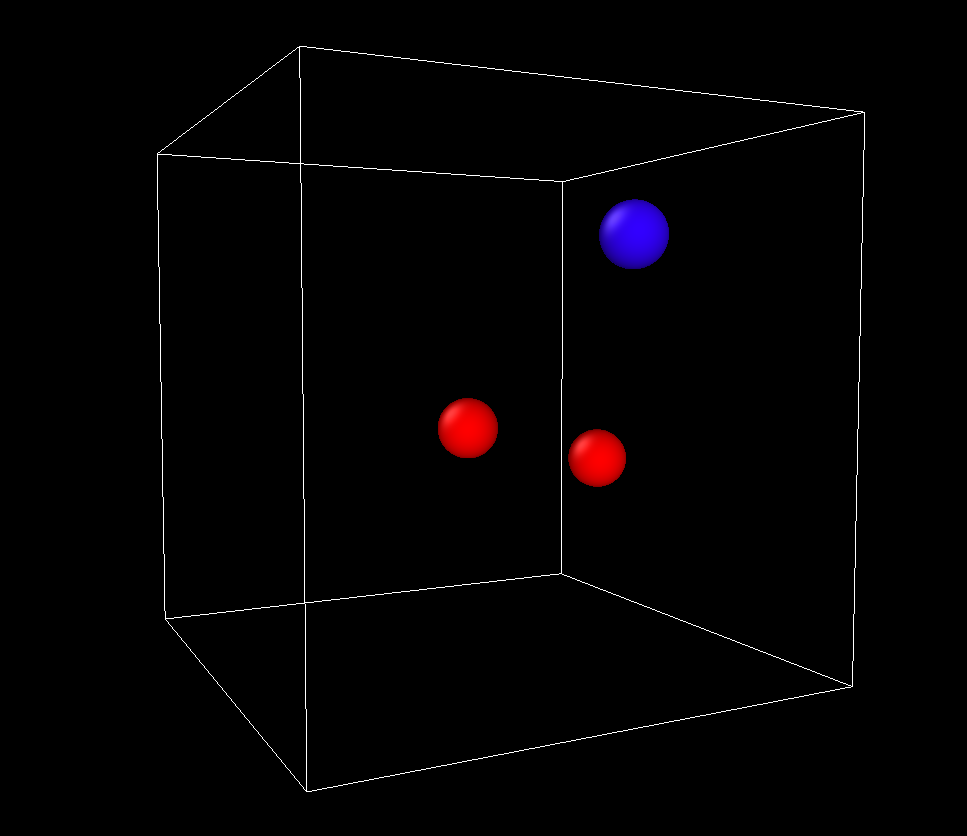
The result, if rendered in Ovito, is two silicon atoms (in red) and one oxygen atom:
About the LAMMPS format
LAMMPS is a molecular dynamics simulation package that is extremely versatile with plenty of interaction potentials and features implemented. However, you may have written some other code involving atoms and found yourself in the position of considering using a standard file format to write atom data to file. In this case, the XYZ-format has likely passed your mind, but because this is a ASCII-based text format, it is slow to read and write. This format also lacks standardized headers for information such as the system boundaries - causing visualizers like Ovito to have to guess for the right boundaries in your system.
The LAMMPS format is binary, so it is way faster than the XYZ-format, and it also carries with it information about the system, which helps Ovito render everything properly.
The LAMMPS file format consists of an header and column-based data stored as doubles. The header consists of the following:
- 1 × 32-bit integer holding the current time step
- 1 × 32-bit integer holding the total number of atoms
- 6 × 64-bit doubles holding the system boundaries
- 3 × 64-bit doubles holding the shearing of the boundaries
- 1 × 32-bit integer holding the number of columns
- 1 × 32-bit integer holding the number of chunks
- 1 × 32-bit integer holding the chunk length, N atoms × n columns.
- (N atoms × n columns) × 64 -bit doubles holding the values of all the atom data
What is stored in the columns?
The 32-bit integer holding the number of columns in the header tells how much data will be stored for each atom. For instance the atom type, 3 position components, 3 velocity components would need 1 + 3 + 3 = 7 columns. The meaning of the data in the columns will have to be interpreted when you open the file in Ovito or any other program, because the LAMMPS format contains no information about the contents of the columns.
Note that all data will have to be stored as 64-bit doubles, even though they might be integers in your application. So you will have to do the conversion to doubles before storing the data to file!
What are chunks?
You don’t really have to care about this. Just store everything as 1 chunk and set the chunk length to the number of columns times the number of atoms (N atoms × n columns).
I think the idea of chunks is used when you build your file with data from multiple processors or split the file up in a logical way to make writing and reading easier for LAMMPS or other applications, but I’m not sure.
Get to the code already
You’ll find an up-to-date example of this code on Github, but I’ve included it below as well for reference:
[cpp]#include <iostream>
#include <armadillo>
#include <fstream>
using namespace std;
using namespace arma;
void writeLammpsFile() {
ivec atomTypes;
atomTypes << 14 << 8 << 14; // Silicon, oxygen,
silicon
cout << atomTypes << endl;
mat positions;
positions << 1.0 << 2.5 << -1.5 << endr
<< 4.0 << -5.0 << 6.0 << endr
<< 7.0 << 2.0 << -3.0 << endr;
cout << positions << endl;
ofstream lammpsFile(“outfile.lmp”, ios::out | ios::binary);
// Set up data for the timestep of this file
int currentTimeStep = 0;
int nAtomsTotal = positions.n_rows;
// The system boundaries
double xMin = -10.0;
double xMax = 10.0;
double yMin = -10.0;
double yMax = 10.0;
double zMin = -10.0;
double zMax = 10.0;
// Shearing is zero unless the system boundaries are sheared (yes that’s
“sheared”,
// not “shared”)
double xShear = 0.0;
double yShear = 0.0;
double zShear = 0.0;
// nColumns is the number of data types you want to write. In our case
we want to
// write four - the atom type and the x, y and z components of the
position.
// If you want velocities, forces, etc., just add more columns and write
more data.
int nColumns = 1 + 3;
// We could divide the data into chunks by the LAMMPS file format, but
we don’t - i.e. only
// use one chunk. The chunk length is then the same as the number of
atoms times the number
// of columns.
int nChunks = 1;
int chunkLength = nAtomsTotal * nColumns;
// Write all the above to the lammps file
lammpsFile.write(reinterpret_cast<const
char*>(¤tTimeStep), sizeof(int));
lammpsFile.write(reinterpret_cast<const char*>(&nAtomsTotal),
sizeof(int));
lammpsFile.write(reinterpret_cast<const char*>(&xMin),
sizeof(double));
lammpsFile.write(reinterpret_cast<const char*>(&xMax),
sizeof(double));
lammpsFile.write(reinterpret_cast<const char*>(&yMin),
sizeof(double));
lammpsFile.write(reinterpret_cast<const char*>(&yMax),
sizeof(double));
lammpsFile.write(reinterpret_cast<const char*>(&zMin),
sizeof(double));
lammpsFile.write(reinterpret_cast<const char*>(&zMax),
sizeof(double));
lammpsFile.write(reinterpret_cast<const char*>(&xShear),
sizeof(double));
lammpsFile.write(reinterpret_cast<const char*>(&yShear),
sizeof(double));
lammpsFile.write(reinterpret_cast<const char*>(&zShear),
sizeof(double));
lammpsFile.write(reinterpret_cast<const char*>(&nColumns),
sizeof(int));
lammpsFile.write(reinterpret_cast<const char*>(&nChunks),
sizeof(int));
lammpsFile.write(reinterpret_cast<const char*>(&chunkLength),
sizeof(int));
// Write all the data for each atom to file
for(uint i = 0; i < positions.n_rows; i++) {
// IMPORTANT: Even though atom numbers are usually integers, they must
be written
// as double according to the LAMMPS standard.
double atomType = atomTypes(i);
lammpsFile.write(reinterpret_cast<const char*>(&atomType),
sizeof(double));
// Write the x, y and z-components
lammpsFile.write(reinterpret_cast<const char*>(&positions(i,0)),
sizeof(double));
lammpsFile.write(reinterpret_cast<const char*>(&positions(i,1)),
sizeof(double));
lammpsFile.write(reinterpret_cast<const char*>(&positions(i,2)),
sizeof(double));
}
lammpsFile.close();
}
int main()
{
writeLammpsFile();
return 0;
}
[/cpp]